




合作客户/
拜耳公司 |
同济大学 |
联合大学 |
美国保洁 |
美国强生 |
瑞士罗氏 |






相关新闻Info
-
> 3种助剂对螺虫乙酯和联苯菊酯药液表面张力、金钗石斛菲盾蚧防治效果的影响(二)
> 铂金板法测定不同浓度、温度、表面活性剂对氨水表面张力值(二)
> 为什么钢针会漂浮在水面上?
> 超微量天平应用案例:铅试金富集称量法测定含铜物料中金和银含量
> 肺内液表面张力的作用、临床意义及测量方法(二)
> 沥青质及其亚组分与烷基苯磺酸钠水溶液在降低IFT中的协同机理(一)
> 一套低温、高压悬滴法表面张力实验测量系统实践效果(一)
> 不同矿浆浓度、粒度、伴生矿物、捕收剂和起泡剂对矿浆表面张力的影响(一)
> 人胰岛素的朗缪尔单分子层膜的表面化学和光谱学性质——摘要、介绍
> 低界面张力纳米流体提高低渗透油藏压裂渗吸速率和采收率(三)
推荐新闻Info
-
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(三)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(二)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(一)
> 表面张力修正系数与蒸汽进口参数的相关性
> 表面张力修正系数与蒸汽膨胀速率的相关性
> 蒸汽自发凝结数值模拟中液滴表面张力修正系数的确定方法(一)
> 融合小波分析的神经网络模型在CO₂-水界面张力预测中的优势与应用
> 表面张力——高精度玩具图案转印的“隐形基石”
> 表面张力均衡在除尘滤袋中的关键作用与革新
> 超低界面张力下重油-水两相垂直流动型态实验研究与图版预测(四)
温度、盐对辛基酚聚氧乙烯醚磺酸盐的油-水界面行为的影响(一)
来源:化工学报 浏览 1151 次 发布时间:2024-11-07
摘要:采用分子动力学模拟(MD)的方法在分子层面上考察辛基酚聚氧乙烯醚磺酸盐(OPES)在油-水界面的界面行为。模拟结果表明:辛基酚聚氧乙烯醚磺酸盐可以大幅降低油-水界面的界面张力,在OPES浓度达到饱和浓度时,系统界面张力仅为3.85 mN·m-1;OPES中磺酸基是主要亲水基团,具有良好的亲水性;温度在318~373 K时,界面张力由24.63 mN·m-1下降到17.43 mN·m-1,这说明OPES具有良好的抗高温性能;当Na+浓度在1%~5%的环境下OPES性质稳定,界面张力仅有4.47 mN·m-1的小幅增加,因此OPES具有良好的耐盐性,并且其对Na+的耐盐性能好于对Ca2+的耐盐性。
在三次采油中,为提高原油采收率,经常利用表面活性剂来降低油水界面张力,目前国内部分油田综合含水量已高达90%,单独的阴离子、非离子型表面活性剂已经不能满足当前的采油要求,阴非离子型表面活性剂作为一种同时有非离子及阴离子表面活性剂优点的两性表面活性剂对于目前日益严苛的采油环境的适应性更强。本文研究的辛基酚聚氧乙烯醚磺酸盐(OPES)是一种具有优良的乳化、耐温、耐盐性能的阴非两性表面活性剂,它已经作为分散剂、润湿剂、乳化剂、洗涤剂等被广泛地应用于石油、日化、纺织等领域。
分子动力学模拟主要是利用牛顿力学来模拟分子的运动,从不同状态下的体系抽取样本进行构型积分并以此为基础计算体系的热力学量等宏观性质。从20世纪90年代后期,人们开始利用计算机模拟研究表面活性剂的性能,它可以将真实环境中的实验现象在分子层面进行解释。对液液界面的研究作为分子动力学模拟的重要研究方向之一近年来受到广泛的关注和报道,如Jang等利用MD模拟了苯磺酸基在不同位置时十六烷基苯磺酸盐的界面张力等界面性能。Wardle等考察了表面活性剂对无机盐、水和正己醇构成的混合物中钠离子迁移的影响。陈贻建等用MD模拟方法对表面活性剂在气-液、固-液、液-液界面的自组装现象进行深刻解释分析。因此利用MD方法研究表面活性剂的界面张力、抗温、抗盐等界面性能具有重要意义。国内对于应用分子动力学模拟来研究表面活性剂性能的起步较晚,特别是对具有耐温、耐盐性能的表面活性剂的研究较少,本文通过分子动力学模拟来研究辛基酚聚氧乙烯醚磺酸盐的油-水界面行为、抗温、抗盐性能,可为实际实验提供较为准确的指导。
1、分子动力学模拟的模型选择与模拟方法
20世纪80年代以来,人们相继研发出可以适合不同环境的力场,如GROMOS、OPLS、AMBER、CHARMM等。本文选择Gromacs中GROMOS53a6力场,以辛基酚聚氧乙烯醚磺酸盐为研究对象进行模型构建。
分子的物理化学性质由其分子结构决定,因此合理的分子结构以及准确的原子电荷是模拟准确性的基础保证。首先要对模拟对象用GAMESS(US)进行结构优化,然后利用Kollman-Singh方法计算电荷,另外如果分子内存在对称结构还需进行电荷平均化来保证电荷分配的合理性。由于本文采用联合原子力场,因此还要去除sp3杂化。图1为优化后的OPES分子结构以及电荷分布,图中绿色小球为碳原子,白色小球为氢原子,红色小球为氧原子,黄色小球为硫原子。
在进行分子动力学模拟之前构建出合理的力场是极为重要的工作。本文通过Autom-ated Topology Builder(ATB)and repository生成的GROMOS系列力场参数,利用现有的数据库以及量子化学进行计算,同时它可以充分考虑到分子中的对称结构,使其反映出的分子性质及参数更为精确。但ATB只能处理原子数小于40的分子,对于分子数大于40的分子结构需进行拆分。在获取准确的电荷及键参数之后利用packmol程序定向排列分子将其堆砌成立方体结构。此外,本文选取的油-水界面需使表面活性剂平均分布在水相两侧,亲水基靠近水相,疏水基靠向油相。图2为初始状态下体系截图,其中中间红色部分为水分子,左右两侧蓝色部分为癸烷分子,油水中间即OPES分子。
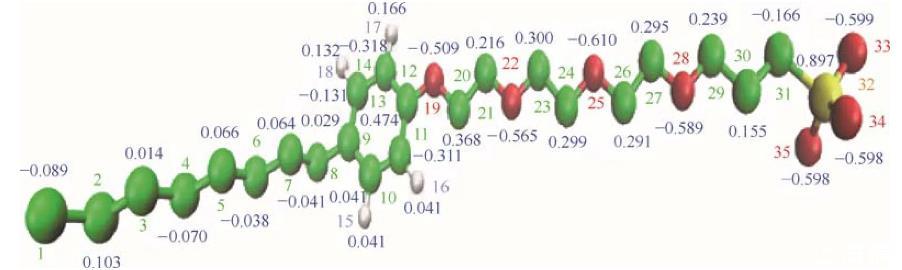
图1辛基酚聚氧乙烯醚磺酸盐的分子结构以及电荷分布
本文中所有体系所堆砌的盒子均为5 nm×5 nm×17.5 nm长方体,并在x、y、z方向选择周期性边界条件。系综选择NPT(等粒子等温等压系综),初始压力为1.01325×105Pa,水模型使用SPC(simple point charge),温度采用Nose-Hoover热浴法,压力采用Parrinello-Rahman压浴法,由于模拟过程中系统为等压变化所以本文模拟的所有系统最终压力值均在1.0081×105~1.0178×105Pa之间。在体系能量最小化后,先进行100 ps的NVT模拟,使体系升温到300K并在此温度下产生初速度,再进行1 ns的NPT模拟使体系密度达到合理状态,再进行12 ns的NPT模拟,控温及控压的弛豫时间为0.5、4.0 ps,积分步长为2 fs,在模拟过程中添加适当的阴阳离子保持体系为电中性。

图2初始状态下体系截图