




合作客户/
拜耳公司 |
同济大学 |
联合大学 |
美国保洁 |
美国强生 |
瑞士罗氏 |






相关新闻Info
推荐新闻Info
-
> 偏硼酸钠复配表面活性剂用于桩西高钙镁油藏超低界面张力驱油体系研究
> 桩西原油与耐垢碱/表面活性剂复合体系的动态界面张力行为
> APTES在绢云母表面的周期性自组装特性
> APTES/乙醇溶液处理与绢云母粉末表面张力测定
> 绢云母粉末表面APTES自组装及其张力变化
> 碱是如何影响重烷基苯磺酸盐体系的界面张力的?(二)
> 碱是如何影响重烷基苯磺酸盐体系的界面张力的?(一)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(三)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(二)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(一)
二糖类的双子表面活性剂性质研究:结果和讨论、结论、致谢!
来源:上海谓载 浏览 2072 次 发布时间:2022-02-24
结果和讨论
方案2和方案3以及实验部分详述了我们获得A系列和B系列海藻糖双子的方法。另外几条评论有助于美化这一信息。B型海藻糖双子最明显的先验途径是用长链叔胺直接取代2的甲苯磺酰化基团。尽管沿着这些路线进行了多次尝试,但只获得了分解产物或起始材料。然而,如方案2所示,我们成功地用叠氮化合物取代了2的两个甲苯磺酰化物,得到了3。此处采用文献程序28,但出于安全考虑,HMPA被DMPU取代作为溶剂。然后,双叠氮化物3经过Staudinger反应25,29,在一个步骤中得到长链双酰胺4a-g,而该长链双酰胺4a-g只需脱保护即可获得a系列双子。在方案3中,脱保护糖5的叠氮化物还原将化合物转化为二伯胺盐酸盐6。长链醛与6和吡啶硼烷络合物30的还原胺化得到海藻糖衍生物7。使用NaBH3-CN作为还原剂,该反应的产率很低。在用甲基碘化物将7甲基化,然后进行离子交换(以促进纯化和提高溶解性)之后,我们得到了B系列海藻糖双子。

必须时刻警惕杂质的存在及其对表面活性剂性质的实质性影响。(可以说,胶体化学家中永远存在不纯的思想。)在我们的案例中,方案2中的前体4a-g在脱保护到最终的A系列双子之前通过柱层析纯化。后者通过1H和13C NMR、高分辨率FAB-MS和元素分析进行了表征。在大多数情况下,C、H和N的实验百分比与理论一致,优于0.30%的绝对值。对于两个系列B双子,在C、H、N和Cl元素分析中也发现了类似的良好一致性。表面张力数据从未显示出杂质的特殊“下降”。
A系列海藻糖双子座
我们的第一个惊喜是发现A系列海藻糖双子座不溶于水。例如,A-12、A-14和A-16(指链长的数字)在1毫升温水中旋转2毫克时会留下沉淀物。即使在70°C的温度下,A-10也会在这种处理过程中形成浑浊的悬浮液。A-8在室温下的可溶浓度仅为1-2 mM(根据动态光散射,在该浓度下,会形成胶束大小的聚集体)。天真地说,人们可能会认为a-8的溶解度大得多,因为该分子由六个羟基和两个酰胺组成。尽管相关的6-氨基-6-脱氧己糖衍生物需要加热才能溶解,25许多烷基化单糖(例如,辛基葡萄糖苷和甲基6-O-(N-庚基羰基)-R-D-吡喃葡萄糖苷或下面的“Hecameg”)是水溶性的。然而,A-8并没有类似的天赋。这里的教训是,用一句老生常谈的话来说,我们的双子座比其各个部分的总和要多(或少)。不管老生常谈与否,双子座的独特属性都取决于它。
溶解度是一个常见且经常被忽略的参数,难以测量,甚至更难理解。由于强大的晶体力,固体可能不溶于水,即使有X射线数据,晶体力的性质也常常模糊不清。在我们的例子中,含有酰胺的分子内和分子间氢键可能有助于固态的热力学稳定性。不溶性也可能源于溶液中不利的溶剂化和聚集效应。A系列海藻糖双子可能会抵抗溶解,因为它们无法以与相应单链段可能的方式形成亲水胶束聚集体。当考虑到双子座的两条链位于双糖的远端,分子内链/链相互作用部分受损时,这种推测就变得可信了。链关联的主题将在后面讨论。
A系列海藻糖双子的水不溶性使我们能够将其作为单分子膜放置在水亚相上。31-33借助表面天平,双子座单分子膜被可移动屏障缓慢压缩,在此过程中,监测膜压力和面积。由此产生的“压力-面积等温线”揭示了双子分子在单分子膜中的堆积行为。图1中给出了A-12和A-18的代表性等温线,其中纵坐标是施加在单层上的表面压力(单位为mN/m或达因/cm),而横坐标是在给定压力下膜中每个分子的面积(单位为Å2)。
图1显示了约120Å2的A-12和A-18(以及此处未绘制的中间链长度)的剥离区域(即,由于分子间接触,等温线偏离基线的区域)。通过校准,烷基糖苷的平均面积为49Å2,34,35,而烃链的分子横截面约为20Å2。33图l由A-12和A-18几乎相同的等温线支持的最简单解释是,海藻糖间隔基而不是其碳氢化合物取代基主导界面填充。显然,烷基从空气/水界面投射到空气中,对薄膜的几何要求几乎没有贡献。尽管如此,链间的分子间范德华力稳定了薄膜。这是因为崩塌压力(即单分子膜最终“断裂”时的高压)与链长正相关,例如A-18(53 mN/m)和A-12(47 mN/m)。

图1。使用表面天平获得A-12和A-18的压力-面积等温线,显示出在23°C下约120Å2的提升面积。图为每种表面活性剂10次扫描的平均值。
当固体A-10和A-12双子座浸入水中,并在3-4小时后通过光学显微镜进行检查时,囊泡结构从表面发出(图2)。当1毫米A-10的乳白色悬浮液通过100纳米聚碳酸酯过滤器多次挤出时,根据动态光散射,形成了直径为100纳米的囊泡群。另一方面,固体A-l4、A-16和A-18的水合作用在室温下不会产生囊泡。A-10/A-12与其更高同系物之间的行为差异表明,相态在其中发挥了作用。因此,已知短链脂质在室温下存在于流体“液晶”相中,而长链脂质更喜欢高度有序的“凝胶”相。36凝胶到液晶的转化发生在转变温度Tm。在Tm上方,水可以进入“松散”液晶晶格的双层中,从而在水合时形成囊泡和小管。即使在室温下,A-18U(即9位顺式不饱和的A-18)的水合作用也会产生大量的囊泡和小管(图2),正如人们从一种Tm无疑远低于室温的化合物中所预期的那样。
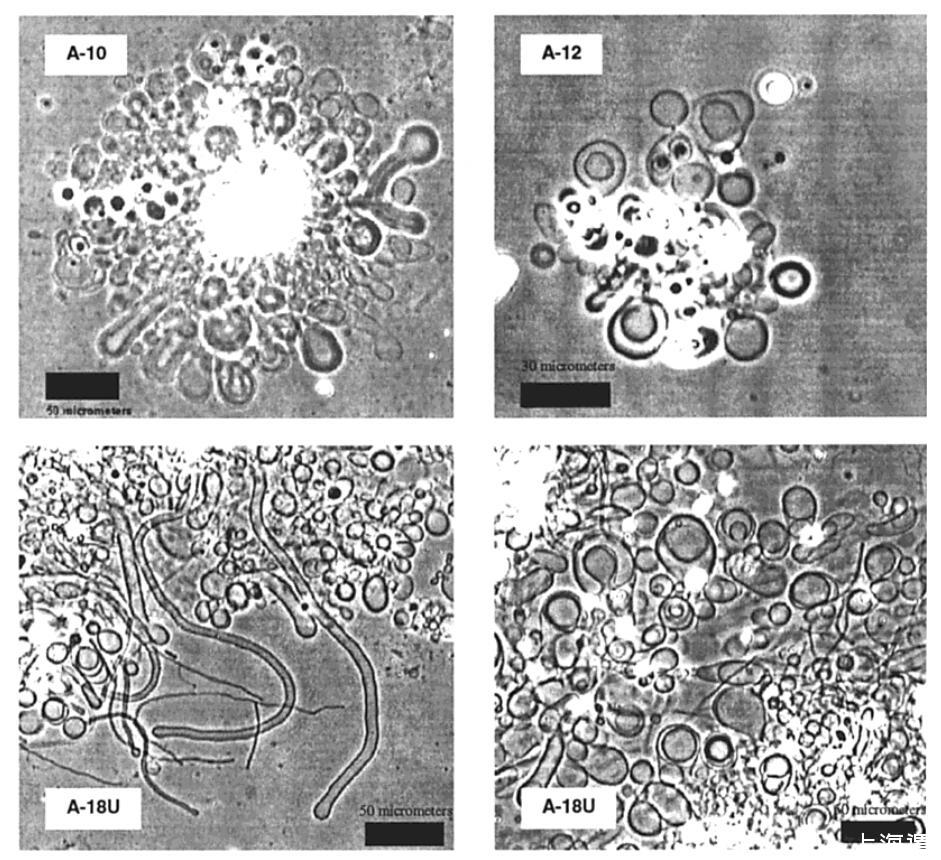
图2。A-10(左上)、A-12(右上)和A-18U(下)在23°C下水合3-4小时后形成的囊泡和管状结构的相衬显微照片。在45°C下培养的A-14和A-16样品中观察到类似结构。
差示扫描量热法扫描是通过在量热计中以10°C/h加热2 mg/mL gemini悬浮液获得的,其吸热峰分别对应于a-14、a-16和a-18的Tm值33、48和61°C(图3)。只有A-l8有一个尖峰,表明在紧密组织的分子阵列中协同熔化。当固体A-14和A-16在45°C下水合时,观察到与图2中类似的光学显微镜图像,而不是在室温下缺乏双层形成。

图3。通过差示扫描量热法(DSC)在10°C/h的加热速率下获得的双子座A-14、A-16和A-18的2 mg/mL悬浮液的吸热峰。
迄今为止的总结是:A系列海藻糖双子,对于10或更长的链不溶于水,形成单分子膜,其界面区域主要由糖间隔基控制。分子间的链/链相互作用稳定了薄膜。A-8在水中形成小胶束,而水合后,更高的同系物聚集成泡状和管状结构,在光学显微镜下可见。水化性质与凝胶到液晶的转变相一致,事实上,转变温度可以通过量热法确定。因此,海藻糖双子的行为与磷脂和合成双链表面活性剂的行为相似。17,37就好像海藻糖不是双子座的间隔物,而是一个特别大但传统的头群。
值得注意的是,我们的双子座研究利用了许多技术,包括合成、薄膜研究、光散射、显微镜、量热法,以及(我们很快就会看到)张量法、电导法和分子力学。在胶体化学中,一种平衡的观点不能仅仅通过一种方法来实现,而只能通过多种方法之间的一致性来实现。
B系列海藻糖双子座
接下来,我们提出了在海藻糖衍生物上放置阳离子基团以改善其水溶性的想法。必须承认,我们对更易管理的水溶性化合物的前景感到一定程度的高兴。B系列就这样出现了。然而,我们立即面临新的问题。虽然B-12和B-14是作为纯化合物获得的,但B-16和B-18并非如此。后两种Gemini的产率较低,提供了令人满意的光谱数据,但它们的元素分析偏离了理论1-2%(这使我们怀疑存在无机杂质)。无论如何,由于B-16和B-18不符合我们的高纯度标准,我们在注意力上轻视了它们,而偏爱它们的矮表亲。
B-12和B-14确实是水溶性化合物,在临界胶束浓度(CMC)以上形成胶束,动态光散射显示颗粒大小小于5nm。CMC值是通过表面张力-浓度曲线(图4)和电导率-浓度曲线(图5)中的断裂来确定的。38表1给出了结果数据(以及两种常规表面活性剂的数据,用于比较)。如图所示,B-12的CMC值通过张力测量法和电导测量法(这两种方法都是可重复的)得出的结果不一致。这种差异至少部分可以追溯到B-12表面张力的时间依赖性。因此,在10分钟的过程中,B-12水溶液的表面压力增加了10 mN/m,之后趋于稳定。这种动态表面张力过程(之前已经用双子座表面活性剂观察到)13,15,39可能与双子座表面活性剂在空气/水界面组织方面的困难有关。为了解决这个问题,图4中的B-12溶液在表面张力测量之前老化20分钟,从而减少了系统尚未平衡的可能性。尽管采取了这种预防措施,但我们无法确定依赖时间的界面重组是否不会影响B-12图,并导致基于张量和电导的数据之间的差异。当然,后者来源于不受界面事件影响的大块特性,因此更可靠。有趣的是,B-14没有表现出随时间变化的表面张力,两种方法确定的CMC值是合理一致的。也许更长的链既增强了对空气/水界面的亲和力,也增强了从溶液中吸收界面的动力学。
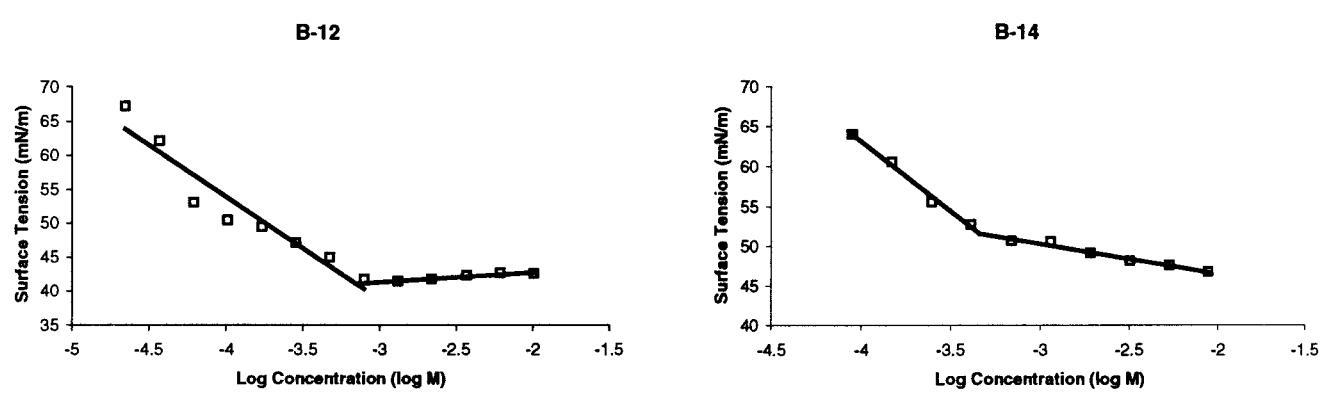
图4。室温下测定B-12和B-14临界胶束浓度的张力测定图。B-14混合后立即进行测量,而B-12样品老化20分钟。
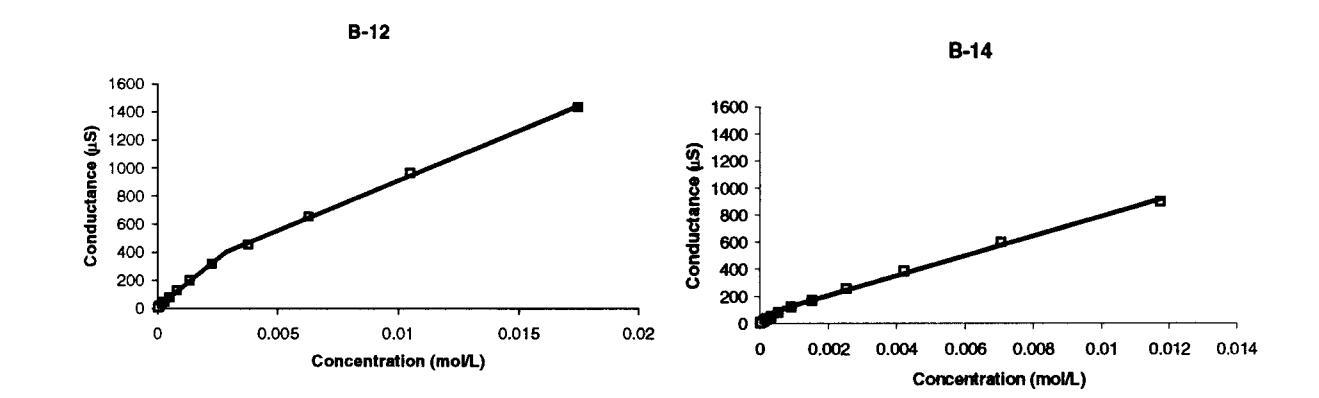
图5。图中给出了B-12和B-14临界胶束浓度,该浓度由电导-浓度曲线中的断裂点确定。每个点代表23°C下的单个电极读数。
表1包含一些显着的比较。从电导数据来看,将B系列中的12个碳链延长至14个碳链,可使CMC降低约4倍(对链长的敏感性略低于传统表面活性剂)。B-12的CMC比单链阳离子表面活性剂十二烷基三甲基氯化铵(DTAC)的CMC小一个数量级。再一次,双子座更容易自我组装的倾向得到了肯定。1,39由于B-12的CMC仅为无糖双子座12-2-12的3倍(即C12H25N+(Me)2C2H4N+-(Me)2C12H25,2Br-),40考虑到其多羟基,糖对胶束化的抑制程度相对较小。胶束表面的分子间氢键41似乎弥补了单体亲水性的增强。

分子模拟
一个核心问题仍然有待解决:为什么A系列双子座与10个或更多碳形成囊泡,而B系列双子座更喜欢胶束状态?为了正确地面对这个问题,有必要寻求分子建模的帮助。因此,我们可以评估形态是否有构象根。
计算首先将海藻糖插入宏模型程序42,并对结构进行琥珀色能量最小化。从使用琥珀色*力场的25000个构象搜索中,我们选择了能量最低的构象,该构象恰好与报告的X射线结构重叠。43,44接下来,该结构中的6-和6′-羟基被乙酰氨基取代。在模拟的水环境中,用AMBER*再次进行了25000次构象搜索。然后乙酰基被十二烷基取代,然后对生成的a-12双子座进行了另一次搜索。结果表明,整个构象由酰胺羰基和相邻葡萄糖环的2-羟基之间的两个分子内氢键控制。氢键导致两条全反式链彼此摆动约95°。A-12的固态红外光谱证实了这种相互作用,该光谱显示1646 cm-1处有羰基带,而Hecameg的羰基带为1700 cm-1,单体葡萄糖类似物的羰基带为1698 cm-1。25由于这种构象会不切实际地排除分子内疏水缔合,而且在任何情况下,羰基氢键位点的溶剂可能会淹没在水中,我们故意不允许-CdO氢键,并重新计算。通过这种方式,我们得到了图6。可以看到,一条链上大约有八个碳与其伙伴链的碳并排排列。N/N距离仅为7.4Å,整体分子形状为“管状”。用另一种方式说,糖的头基延伸的距离不比两条链的宽度大多少,就像人们预期的一种易于堆积成双层的化合物那样。45因此,正如前面从实验数据得出的结论,海藻糖更像是一个统一的头基,而不是一个可以有效隔离链的长间隔基。
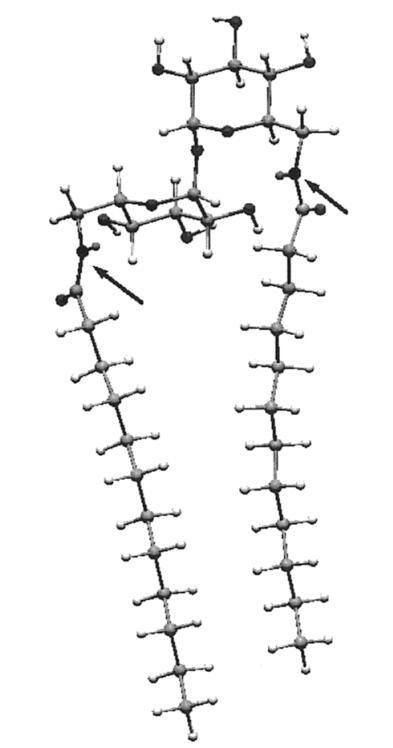
图6。A-12最低能量构象的宏观模型快照,显示了双层形成两亲分子的管状结构特征。箭头表示酰胺氮(N-to-N距离)7.4Å)。
B系列双子座的情况则截然不同(图7)。N/N距离现在为9.3Å,导致B-12分子呈圆锥形而非管状。人们认为,当头基的面积超过链的面积时,就不利于双层的形成,胶束化是自组装的主要方式,这一点在这里很明显。45因此,与a系列相比,B系列的胶束化是构象效应的直接结果,构象效应产生了更大的头基单元。B系列中阳离子头基之间的静电排斥也可能起作用。由于胶束不如双层膜紧密,B系列化合物的阳离子头基之间的排斥力有利于胶束化。然而,静电不可能是全部,因为双链阳离子,如二十二烷基二甲基溴化铵,自组装成双层膜没有问题。17
结论
合成了一系列带有双糖(海藻糖)间隔基的双子表面活性剂,使6-和6′-羟基被长链酰胺(A系列)或长链季铵盐(B系列)取代。两个系列的胶体行为不同。A系列双子座不溶于水,在水中水合或挤压时形成泡状结构。通过动态光散射、显微镜、量热法和分子模拟对这些结构进行了表征。此外,化合物以单层形式沉积在水性亚相上,并用表面天平进行检查。系列B双子座是水溶性的,除了进行分子模拟外,还进行了张力测定和电导测定。实验数据和理论分析一致地定义了新表面活性剂假定的不同形态。由此揭示了形态和结构之间的相关性。
实验段
一般方法。所有试剂均从Aldrich购买。溶剂在4Å分子筛上干燥。在Whatman正相硅胶板上进行TLC,并通过UV或碘可视化观察。熔点用Thomas Hoover毛细管熔点仪测定,未经校正。在Varian INOVA 300或400分光光度计上记录1H NMR和13C NMR光谱。质谱数据来自埃默里大学质谱中心;为此,我们承认使用了NIH和NSF提供的共享仪器。元素分析由佐治亚州诺克罗斯的大西洋显微实验室进行。
将六邻乙酰基-6,6′-二邻甲苯对磺酰-6,6′-二脱氧-r,r-DTR-海藻糖2.28二水合物(10.0 g,26 mmol,52 mmol当量)溶解于140 mL吡啶中。搅拌时缓慢添加25.1 g(130 mmol)对甲苯磺酰氯,溶液颜色从石灰绿变为淡黄色;热量散发出来。反应在35分钟内完成,混合物用140毫升醋酸酐淬火。搅拌一夜后,将所得深色溶液倒在约1.5升冰水上。收集沉淀并从甲醇中再结晶三次,得到3.2 g(14%)的2,为白色固体,mp 162-164°C;点燃。28兆帕170-172摄氏度。
1H NMR(300MHz,CDCl3;C2表示C2和C2′等)δ7.73(d,J)8.1,arom。H邻位到C-SO2,4H),7.34(d,J)8.1 Hz,arom。H与C-CH3,4H正交,5.41(t,J)9.6 Hz,C3,2H),4.92(m,6H),4.10(m,6H),2.44(s,Me of Tos,6H),2.07(s,C2 Ac,6H),2.00(s,C3 Ac 6H),1.98(s,C4 Ac,6H)。13C NMR(CDCl3,75MHz)δ170.09、169.71、145.48、132.56、130.04、128.21、92.96、69.95、69.34、68.71、68.31、67.71、21.83、20.74。FAB-HRMS(M+Li)+:计算909.2133。发现:909.2144。肛门。C38H46O21S2的计算值:C 50.55;h5.14;S 7.10。发现:c50.69;h5.14;S 7.02。
将2,3,4,2′,3′,4′-六氧乙酰基-6,6′-二叠氮-6,6′-二脱氧-r,r-D-海藻糖,3.28加入14.9 g(16.5 mmol,33 mmol当量)二糖基海藻糖衍生物2和30 mL 1,3-二甲基-3,4,5,6-四氢-2-(1H)-嘧啶酮(DMPU)的溶液中,加入2.3 g(35.4 mmol)NaN3。将混合物在90°C的油浴中搅拌6 h。将褐红色反应混合物用水淬火,以产生粘性橙色沉淀物。甲醇的两次再结晶产生6.8g受保护的二氮基海藻糖3(64%),mp 115-117℃;点燃。28MP 114-116摄氏度。
1HNMR(CDCl3,300MHz):δ5.47(t,J)9.9Hz,C3,2H),5.32(d,J)3.6Hz,C1,2H),5.12-4.96(m,C2和C4,4H),4.11-4.06(m,C5,2H),3.40-3.33(m,C6,2H),3.19-3.14(m,C6,2H),2.12(s,C2 Ac,6H),2.06(s,C3 Ac,6H),2.03(s,C4,Ac,6H)。
肛门。计算:C24H32N6O15:C 44.72,H 5.00,N 13.04。发现:C 44.72,H 5.06,N 12.94。
将二嗪3转化为二酰胺4a-g的Staudinger转化的一般程序。通过注射器将适当的酸性氯化物注入3和CH2Cl2的搅拌溶液中。搅拌5分钟后,添加溶解在CH2Cl2中的三苯基膦(PPh3),并在室温下搅拌溶液2天。在最初几分钟内,观察到气泡从反应溶液中冒出(释放N2)。正相薄层色谱(2:1乙酸乙酯:石油醚作为洗脱液)显示反应完成,显示形成了一个非常显着的紫外可见斑点,Rf为0.3,对应于三苯基膦盐副产物。用NaHCO3水溶液(10 g/200 mL)洗涤混合物,并在MgSO4上干燥有机溶液。去除溶剂,通过硅胶柱层析(2:1乙酸乙酯:石油醚)实现纯化。
2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二十八胺基-6,6′-二脱氧-r,r-D-海藻糖,4a。按照上述一般程序,2.1 g(3.3 mmol,6.5 mmol当量)3,50 mL CH2Cl2和2.7 mL(9.8 mmol)辛酰氯,以及2.4 g(9.2 mmol)PPh3与15 mL CH2-Cl2反应。黑色固体被过滤,留下栗色溶液。洗涤和干燥后,对所得棕色糖浆进行色谱分析,得到2.2 g(80%)4a,白色固体,mp 70-72°C。
1H NMR(300 MHz,CDCl3):δ5.67(t,J)5.7 Hz,N上的H,2H),5.46(t,J)9.9 Hz,C3,2H),5.30(d,J)3.6 Hz,C1,2H),4.93-4.84(m,C2和C4,4H),3.83(m,C5,2H),3.58-3。52(dd,J)12.6,6赫兹,C6,2H),3.33-3.24(m,C6,2H)2.21-2.01(m,R CH2,4H),2.07(s,C3 Ac,6H),2.07(s,C4 Ac,6H),2.02(s,C2 Ac,6H),1.62-1.60(m,R CH2,4H),1.28(m,烷基链,16H),0.86(t,J)6赫兹,CH3,6H)。
13C NMR(75 MHz,CDCl3):δ173.25、170.24、169.93、169.85、91.87、70.41、69.87、69.46、69.23、38.98、36.86、31.99、29.57、29.29、25.77、22.92、21.00、20.9.
FAB-LRMS(M+Li)+:851.8
肛门。C40H64N2O17的计算结果:C 56.86,H 7.63,N 3.32。发现:c56.81,h7.71,n3.15。
2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二十二酰胺-6,6′-二脱氧-r,r-D-海藻糖,4b。使用2.1 g(3.3 mmol,6.5 mmol当量)3、50 mL CH2Cl2、1.9 mL(9.0 mmol)癸酰氯和2.2 g(8.4 mmol)PPh3在10 mL CH2-Cl2中的溶液进行反应。通过硅胶柱层析纯化,得到0.86 g(37%)4b,mp 62-66°C。
4.46(t,J)3.57-3.52(dd,J)3.3-3.6、10.10-3.6、10.8赫兹、10.8赫兹、C6、6、6、2、2H、6、6、6、6、6、6、6、6、3、3.57-3.52(dd、J)3.6、3.6、10.8、10.8赫兹、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、6、3、3、3)、3.30-3-3.30-3-3-3.26、3-3.30-3.26、3.26、3.30-3-3.26(3.26、3、3、3、(m,CH2,4H),1.29-1.25(m,烷基链,24小时),0.87(t,J)6.8赫兹,CH3,6小时)。
13C NMR(100 MHz,CDCl3):δ173.45、170.43、170.11、170.04、91.89、70.39、69.84、69.44、69.21、38.90、36.77、32.06、29.67、29.54、29.48、25.287、22.87、01.0.。
FAB-HRMS(M+Li)+:计算907.4991。发现:907.4974。
肛门。C48H80N2O17的计算值:C 58.65,H 8.05,N 3.11。发现:C58。52,H7.93,N3.05。
2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二脱氧卡那酰胺-6,6′-二脱氧,r-D-海藻糖,4c。将3(2.0 g,3.2 mmol,6.4 mmol当量)、50 mL CH2Cl2、2.2 mL(9.8 mmol)月桂酰氯和2.20 g(8.4 mmol)PPh3与10 mL CH2Cl2反应。橙黄糖浆的硅胶层析,然后从己烷中进行重结晶(以去除1H NMR检测到的PPh3盐杂质),得到1.08 g(36%)纯4c,mp 64-68°C。
4.47(t,J)12.9,6.3(dd,J)12.9,6.3赫兹,6.3赫兹,C66,2H),3.33-3.26(m,C6,2H),3.33-3.26(m,C6,2H),3.33-3.26-3.26(m,C6,6,26(m,6,6,2H),3.26(m,6,6,6,2H),2),2,2,2,2.2-2-2-2-2-2.2-2-2-2.1(m,2-2-2-1(m,R,CH2,R,CH2,CH2,2,2),2-2-2-2-1(m,R,2,CH2,2,CH2,2,2,2,2,2,2,2-2,2,2,2,2-2-2(m,CH2,2,2,2 CH2,4H),1.25(m,烷基链,32H),0.88(t,J)6.9 Hz,CH3,6H)。13C NMR(100 MHz,CDCl3):δ173.45、170.42、170.11、170.03、91.89、70.38、69.84、69.43、69.21、38.90、36.77、32.11、29.82、29.71、29.54、25.67、22.89、20.88、20.80、14.33。FAB-HRMS(M+Li)+:计算964.5695。发现:964.5668。肛门。C48H80N2O17+1/2 mol H2O的计算:C 59.67,H 8.45,N 2.90。发现:C 59.33,H 8.23,N 2.81。2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二乙酰氨基-6,6′-二脱氧,r-D-海藻糖,4d。3(2.1 g,3.2 mmol,6.4 mmol当量)、50 mL CH2Cl2、2.4 mL肉豆蔻酰氯(9.0 mmol)和2.3 g PPh3(8.8 mmol)在10 mL CH2Cl2中按照一般程序反应。通过柱层析纯化淡黄色糖浆,得到0.9g(27%)4d,mp 56-60°C。1H NMR(400MHz,CDCl3):δ5.71(t,J)6Hz,N上H,2H),5.46(t,J)10Hz,C3,2H),5.30(d,J)4Hz,C1,2H),4.92-4.84-3.81(m,C5,2H),3.54-3.52(m,C6,2H),3.30-3.26(m,C6,2H),2.20-CH2,4H,2.07(s,C3 Ac,6H),2.07(s,C4 Ac,6H),2.02(s,C2 Ac,6H)1.59(m,CH2,4H),1.29-1.24(m,烷基链,40H),0.87(t,J)6.4赫兹,CH3,6H)。13C NMR(100 MHz,CDCl3):δ173.52、170.43、170.13、170.06、91.89、70.38、69.84、69.42、69.21、38.89、36.77、32.12、29.86、29.73、29.56、25.68、22.89、20.88、20.79、14.33。FAB-HRMS(M+Li)+:计算1019.6243。发现:1019.6274。肛门。C52H88N2O17的计算值:C 61.64,H 8.75,N 2.76。发现:C 61.69,H 8.86,N 2.64。2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二十六烷酰胺-6,6′-二脱氧,r-D-海藻糖,4e。用2.1 g(3.2 mmol,6.4 mmol当量)3,50 mL CH2Cl2,2.7 mL(9.0 mmol)棕榈酰氯和2.4 g(9.1 mmol)PPh3在10 mL CH2Cl2中进行反应。米白色粘性固体的柱层析提供2.9 g(73%)4e作为白色结晶固体,mp 54-57°C。1H NMR(400 MHz,CDCl3):5.68(t,J)6 Hz,N上H,2H,5.46(t,J)9.6 Hz,C3,2H,5.30(d,J)3.6 Hz,C1,2H),4.92-4.85-3.80(m,C5,2H),3.57-3.52(m,C6,2H),3.31-3.26(m,C6),2.20-2.10(m,R CH2,4H),2.07(s,C3 Ac,6H),2.06(s,C4 Ac,6H),2.01(s,C2 Ac,6H)1.59(m,R CH2,4H),1.29-1.24(m,烷基链,48H),0.87(t,J)6.8 Hz,CH3,6H)。13C NMR(100 MHz,CDCl3):δ173.48、170.43、170.11、170.04、91.89、70.38、69.84、69.42、69.21、38.90、36.78、32.12、29.90、29.73、29.57、25.68、22.90、20.88、20.80、14.33。FAB-HRMS(M+Li)+:计算1075.6869。发现:1075.6865。(M+H)+:1069.6787的计算。发现:1069.6749肛门。C56H92N2O17的计算:C 62.90,H 9.05,N 2.62。发现:C 62.80,H 9.01,N 2.50。2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二十八酰胺-6,6′-二脱氧,r-D-海藻糖,4f。按照一般程序,2.2 g(3.4 mmol,6.8 mmol当量)3,50 mL CH2Cl2,1.9 mL(9.0 mmol)硬脂酰氯和2.29 g(8.7 mmol)PPh3与10 mL CH2Cl2反应。4f(1.9 g,50%),mp 53-56°C,通过粘性淡橙色固体的硅胶色谱分离。1HNMR(400MHz,CDCl3):δ5.70(t,J)6Hz,N上的H,2H),5.45(t,J)9.9Hz,C3,2H,5.29(d,J)3.6Hz,C1,2H,4.92-4.86(m,C2和C4,4H),3.82(m,C5,2H),3.60-3.50(m,C6,2H),3.33-3.29(m,C6,2H),2.18-2.16(m,R,CH2,4H),2.06(s,Ac,6H,Ac,2,Ac,Ac,2C,2,C4,6H),2,C4,6H,1.28-1.24(m,烷基链,56H),0.86(t,J)6.4赫兹,CH3,6H)。13C核磁共振(100MHz,CDCl3):173.43、170.41、170.09、170.02、91.87、70.37、69.83、69.42、69.21、38.89、36。76,32.12,29.89,29.73,29.56,25.67,22.88,20.87,20.79,14.33.FAB-HRMS(M+Li)+:计算1131.7495。发现:1131.7443。肛门。C60H104N2O17+1 mol H2O的计算:C 63.02,H 9.34,N 2.45。发现:C 63.03,H 9.21,N 2.50。2,3,4,2′,3′,4′-六邻乙酰基-6,6′-二-(Z)-十八烷基-9-烯酰胺基-6,6′-二脱氧-r,r-D-海藻糖,4g。将3(2.0 g,3.1 mmol,6.2 mmol当量)、50 mL CH2Cl2、2.98 mL(9.0 mmol)油酰氯和2.2 g(0.4 mmol)PPh3与10 mL CH2Cl2反应。通过硅胶柱层析纯化得到0.8 g(23%)4g,呈吸湿性、粘性固体,mp 50-56°C。1H NMR(300 MHz,CDCl3):5.66(t,J)5.6 Hz,N上H,2H,5.48(t,J)10 Hz C3,2H,5.36-5.34(m,CdC,4H),5.32(d,J)3.6 Hz,C1,2H),4.94-4.86(m,C2和C4,4H),3.86-3.82(m,C5,2H),3.58-3.54(m,C6),3.33-3.26(m,C6,2H),2.22-2.20(m,R CH2,4H),2.09(s,C3 Ac,6H),2.08(s,C4 Ac,6H),2.03(s,C2 Ac,6H),2.01(m,CH2 R到CdC,8H),1.63(m,CH2,4H),1.27(m,烷基链,40H),0.89(t,J)6.4 Hz,CH3,6H)。FAB-HRMS(M+Li)+:计算1127.7182。发现:1127.7137。肛门。C60H100N2O17+1/2 mol H2O的计算:C 63.75,H 9.01,N 2.48。发现:C 63.55,H 8.93,N 2.44。泽姆彭条件下脱乙酰化的一般程序。25,46在室温下,将完全保护的表面活性剂在50 mL甲醇和2 mL 0.57 M NaOMe/MeOH溶液中搅拌过夜。添加约1.5 g洗涤过的(丙酮和甲醇)Dowex MR-3混床离子交换树脂,并搅拌混合物30分钟,或直到pH值达到7。过滤中和后的溶液,在减压下去除溶剂,并在真空中干燥所得固体。6,6′-双十八胺-6,6′-二脱氧-r,r-D-海藻糖,A-8。如上所述,对4a(1.5 g)进行脱乙酰化,得到1.0 g(95%)的A-8,mp 100-104°C 1H NMR(300 MHZ,CD3OD):δ5.03(d,J)3.3 Hz,C1,2H),3.85(m,2H),3.74(t,J)9.3 Hz,2H),3.45(m,6H),3.12(t,J)9.3 Hz,2H),2.20(t,J)7.5 Hz,R CH2,4H),1.59(m,CH2,4H),1.30(t,J)烷基链(6H,6H)。13C NMR(75MHz,CD3OD):δ177.23,95.68,74.28,73.48,73.26,72.22,41.44,37.18,33.05,30.51,30.32,27.29,23.87,14.60。FAB-HRMS(M+H)+:计算593.3661。发现:593.3661。肛门。C28H52N2O11的计算值:C 56.74,H 8.84,N 4.73。发现:c56.58,h8.73,n4.69。6,6′-二十二酰胺-6,6′-二脱氧-r,r-D-海藻糖,A-10。4b(0.8g)脱乙酰化以产生0.6g(100%)A-10作为结晶固体,mp 125-128°C.1H NMR(400MHz,CD3OD):δ5.02(d,J)4Hz,C1,2H,3.86-3.83(m,2H),3.73(t,J)8.8Hz,2H),3.45-3.44(m,6H),3.11(t,J)8 Hz,2H),2.20(t,J)6.8Hz,R CH2,4H),1.59(m,CH2,4H),1.45-3.44(m,6H),3.45-3.44(t,6H),3.18(t,6H),248Hz,6H)烷基链,CH3,6H)。13C核磁共振(100MHz,CD3OD):δ177.23,95.69,74.30,73.49,73.28,72.23,37.19,33.22,30.78,30.66,30.55,27.30,23.91,14.62。FAB-HRMS(M+Li)+:计算655.4357。发现:655.4350。肛门。C32H60N2O11+1/2 mol H2O的计算值:C 58.43,H 9.35,N 4.26。发现:C 58.66,H 9.20,N 4.09。6,6′-双脱氧氨基-6,6′-双脱氧-r,r-D-海藻糖,A-12。4c在泽姆彭条件下脱乙酰化得到0.63克(79%)二氨基海藻糖衍生物A-12,mp 168-172℃。1H NMR(400 MHz,CD3OD):δ5.03(d,J)3.3 Hz,C1,2H),3.86-3.84(m,2H),3.74(t,J)9.2 Hz,2H),3.45-3.42(m,6H),3.11(t,J)10 Hz,2H),2.19(t,J)7.6 Hz,R CH2,4H),1.59(m,CH2,4H),1.28(m,烷基链,32H),0.88(t,J)6.4 Hz,CH3,6H)。13C核磁共振(100MHz,CD3OD):δ177.20,95.66,74.29,73.48,73.27,72.21,41.45,37.18,33.25,30.96,30.92,30.81,30.66,30.56,27.29,23.91,14.63。FAB-HRMS(M+Li)+:计算711.4983。发现:711.4952。肛门。C36H68N2O11的计算值:C 61.34,H 9.72,N 3.97。发现:C61.21,H9.67,N3.90。6,6′-二乙酰氨基-6,6′-二脱氧-r,r-D-海藻糖,A-14。将0.9 g 4d脱乙酰化,然后在真空中干燥,得到0.6 g(88%)的A-14,mp 174-179℃。1H NMR(400 MHz,CD3OD):δ5.05(d,J)3.6 Hz,C1,2H,3.86-3.83(m,2H),3.73(t,J)9.2 Hz,2H,3.45-3.41(m,6H),3.11(t,J)9.2 Hz,2H),2.19(t,J)7.2 Hz,R CH2,4H),1.58(m,4H),1-烷基链,0.88(t,J)6.8 Hz,CH3,6H)。13C核磁共振(100MHz,CD3OD):δ177.22,95.66,74.30,73.49,73.29,72.22,41.46,37.20,33.26,30.97,30.83,30.67,30.57,27.31,23.92,14.63。FAB-HRMS(M+Li)+:计算767.5609。发现:767.5609。肛门。C40H72N2O11的计算:C 63.13,H 10.07,N 3.68。发现:C 63.00,H 10.06,N 3.55。6,6′-二十六烷酰胺-6,6′-二脱氧-r,r-D-海藻糖,A-16。4e(2.9 g)在Ze'mpen条件下脱保护,以提供1.8 g(86%)结晶和吸湿性A-16,mp 189-190°C。1H NMR(400 MHz,CD3OD):δ5.02(d,J)3.6 Hz,C1,2H),3.85-3.83(m,2H),3.74(t,J)9.2 Hz,2H),3.45-3.41(m,6H),3.11(t,J)9.2 Hz,2H),2.19(t,J)7.6 Hz,4H),1.58(m,4H),1.29-1.22(m,烷基链,48小时),0.88(t,J)6.4赫兹,CH3,6小时)。13C-NMR(100mhz,CD3OD):δ177.18,95.64,74.30,73.48,73.28,7221,41.45,37.21,33.26,31.00,30.84,30.67,30.57,27.31,23.92,14.64.FAB-HRMS(M+Li)+:计算823.6235。发现:823.6263。肛门。计算:C44H76N2O11+1/2 mol H2O:C 63.97,H10。37,N 3.39。发现:C64.13,H10.32,N3.30。6,6′-二十八酰胺-6,6′-二脱氧-r,r-D-海藻糖,A-18。当按照所述方案脱乙酰1.9 g 4f时,观察到脱保护表面活性剂以细白色固体形式从溶液中沉淀。真空干燥收集的白色固体,发现1.0g(68%)的A-18是一种精细的白色吸湿固体,熔点192-196℃。1H NMR:(400MHz,1:1Cd3OD:CDCl3,CHD2OD峰用作参考):δ5.17(d,J)4Hz,C1,2H),3.98(m,2H),3.90(t,J)9.2Hz,2H),3.70-3.61(m,4H),3.53-3.48(m,2H),3.29(t,J)9.2,2H),2.36(t,J)7.6Hz,R CH2,4H),1.77-1.73(m,CH2,4H),1.45-1.41(m,烷基链,56H),1.03(t,J)6.4,CH3,6H)。13C NMR(100 MHz,1:1 CD3OD:CDCl3,CDCl3峰用作参考):δ176.74,95.06,73.51,72.76,72.45,71.45,40.96,36.93,32.66,30.42,30.27,30.08,26.69,23.38,14.49。FAB-HRMS(M+Li)+:计算879.6861。发现:879.6832。肛门。C48H92N2O11+1/2 mol H2O的计算:C 65.35,H 10.62,N 3.18。发现:C 65.25,10.40,3.18。6,6′-二-(Z)-十八碳-9-烯酰胺-6,6′-二脱氧-r,r-D-海藻糖,A-18U。0.8克4g的0.8克的脱乙酰基0.8克的4g的脱乙酰乙酰化的0.8克(100%100%)的4g-18U,mp 154-158℃1HNMR(400兆赫,CD3OD)1HNMR(400兆赫,CD3OD)1HNMR(400兆赫,CD3OD):δ5.32(t,J)5.32(t,J)5.2赫兹,5.2赫兹,4H,4H,5.2,5.2赫兹,4H,5.2,4H,5.2,5.2,5.2,5.2,5.02(d,5.02(d,3.02(d,J,J)5.0(d,J)5.8(d,J)5.8)5.2)5.5.2.2.2,4.2,4.2,4赫兹,4赫兹,4赫兹,4赫兹,4赫兹,4,4,40H),0.88(t,J)6.4 Hz,CH3,6H)。13C NMR(100 MHz,CD3OD):δ177.18、131.04、130.96、95.68、74.31、73.49、73.29、72.22、41.47、37.19、33.25、31.02、30.80、30.64、30.56、30.43、28.31、27.31、23.92、14.64。FAB-HRMS(M+Li)+:计算875.6548。发现:875.6555。肛门。C48H88N2O11+1/2 mol H2O的计算:C 65.65,H 10.21,N 3.19。发现:C65.36,H9.98,N3.16。6,6′-二叠氮-6,6′-二脱氧-r,r-D-海藻糖,5.28六邻乙酰化二叠氮-海藻糖3(2.8 g,4.3 mmol),在泽姆彭条件下脱乙酰化,得到1.6 g(94%)脱乙酰基二叠氮5,mp 200-204℃(含dec);点燃。28MP 209-211°C晶圆厂级轻稀土金属(M+Li)+:399.32;(M+2Li-H)+:405.33;(M+3Li-2H)+:411.35。将6,6′-二氨基-6,6′-二脱氧-r,r-D-海藻糖(二HCl盐),6.47脱乙酰二嗪5(6.7 g,17.0 mmol,34 mmol当量)放入120 mL二氧六环和24 mL甲醇的溶液中。添加PPh3(36.1 g,137.8 mmol),并在N2下搅拌溶液一小时。引入NH4OH(31 mL,30%水溶液),并使混合物在N2下搅拌过夜。通过旋转蒸发从白色固体悬浮液中去除溶剂。在真空中干燥5小时后,用150 mL毫Q水(18 M)研磨固体Ω‚cm阻力),并通过小心添加0.1 M HCl将滤液调节至pH 3。过滤残留的PPh3和Ph3PO,并用Milli-Q水研磨。用甲苯洗涤水滤液,然后冷冻干燥,得到6.5 g白色固体,在200°C下分解。如作者所述,试图从MeOH中再结晶47在我们手中并没有得到纯物质。我们发现一些所需的二胺盐被甲醇滤液洗掉了。1H NMR仅显示二氨基海藻糖盐酸盐6的峰。因此,假设杂质是无机盐,我们选择使用粗质子化二胺,无需进一步纯化。1HNMR(400MHz,CD3OD):δ5.3(d,J)3.6Hz,C1,2H),4.08(t,J)9.2Hz,2H,3.90(t,J)9.2Hz,2H,3.77-3.72(m,J)3.6,2H,3.51-3.41(m,4H),3.38-3.20(m,2H)。FAB-HRMS(M-HCl-Cl)+:计算341.1560。发现:341.1573。合成海藻糖双子B-12和B-14的一般程序。将分子筛(4Å)粉碎,用甲醇洗涤,然后在110°C烘箱中干燥过夜。向装有6个和粉碎分子筛的干燥烧瓶中添加甲醇和适当的醛。通过注射器将吡啶硼烷络合物注入烧瓶中,并在N2下搅拌该混合物过夜。反应检查包括用10毫升6 N HCl搅拌溶液一小时,用1 M NaOH将pH值调节到14,并用乙醚萃取混合物。从合并的有机相中去除乙醚并在真空中干燥过夜后,所得橙色残渣用甲醇研磨,以分离任何筛子颗粒。MeOH的旋转蒸发留下了一种玻璃状材料,在K2CO3为碱、MeOH为溶剂的情况下与MeI进行季铵化。将混合物搅拌一夜,用隔膜盖和针插入通风孔。通过干燥混合物分离出的白色固体溶解在Milli-Q纯化水中(18MΩ‚cm抗性),并用CHCl3提取。从组合CHCl3相旋转蒸发溶剂,然后从丙酮结晶。过滤混合物得到白色固体,随后在MeOH中用Dowex(Cl-型)离子交换树脂搅拌过夜。同时,通过将丙酮滤液通过离子交换柱收集额外产物;适当的部分用树脂搅拌一夜,以确保完全的离子交换。在用脱色木炭处理后,将含有树脂的混合物过滤、干燥,然后在丙酮中加热。双生二铵以白色结晶固体的形式通过过滤收集。6,6′-N,N′-二十二烷基二氨基-6,6′-二脱氧-r,r-D-海藻糖,7a。按照上述一般程序,反应0.43 g粉碎的4Å分子筛、40 mL甲醇、2.25 mL(10.19 mmol)十二醛、2.04 g粗二胺二盐酸盐6和0.80 mL(7.92 mmol)吡啶硼烷络合物。检查后,分离出2.06 g粗7a。1hnmr(CD3OD,400mhz):δ5.06(d,J)3.6hz,2H),3.89-3.88(m,2H),3.52-3.46(m,2H),3.10(t,J)9.6hz,2H),2.97-2.93(m,2H),2.68-2.59(m,4H),1.50(t,J)6.4hz,CH2,4H),1.27(m,52H),0.87(t,J)6.4hz,CH3,6H)。FAB-HRMS(M+Li)+:计算683.5398。发现:683.5377。(M+H)+:677.5316的计算。发现:677.5300。6,6′-N,N′-tetramethyldododecyldiamonium-6,6′-dideoxy-r,r-Dtrehalose dichloride,B-12。将上述产物与0.91 g(6.58 mmol)K2CO3和1.5 mL(24.1 mmol)MeI进行季铵化,然后采用上述离子交换和木炭方案,得到1.29 g(32%来自粗二胺二盐酸盐6)B-12,mp>220°C(含dec)。1hnmr(400mhz,CD3OD):δ5.11(d,J)3.9hz,2H),4.49-4.30(m,2H),3.82-3.69(m,4H),3.56-3.52(m,2H),3.49-3.41(m,4H),3.31(m,rch2,4H),3.16-3.10(m,12h),1.79(m,CH2,4H),1.37-1.30(m,36H),0.90(t,J)6hz,CH3,6H)。13C核磁共振(100MHz,CD3OD):δ99.05,74.74,72.95,72.69,69.02,68.82,66.75,66.49,54.97,52.67,33.22,30.91,30.84,30.75,30.63,30.50,27.60,23.88,23.69,14.61。FAB-HRMS(M-Cl)+:计算769.5709。发现:769.5709。肛门。C40H82Cl2N2O9的计算:C 59.61,H 10.25,Cl 8.80,N 3.48。发现:C 59.35,H 10.31,Cl 8.83,N 3.51。6,6′-N,N′-四甲基二乙基二铵-6,6′-二脱氧-r,r-D-二氯化海藻糖,B-14。将粗品6(2.07 g)、0.44 g粉碎分子筛、25 mL甲醇、2.48 g(11.68 mmol)溶于25 mL CHCl3的十四醛和1 mL(9.9 mmol)吡啶硼烷络合物在室温下搅拌过夜,然后在回流下搅拌30 min。加工后,2.92 g粗品7b,用1.21 g(8.75 mmol)K2CO3和2.1 mL(33.73 mmol)MeI进行季铵化。如上所述进行萃取、研磨和离子交换程序后,将0.6 g(粗品6中的14%)的B-14分离为白色固体,mp>200°C(含dec)。1hnmr(400mhz,CD3OD):δ5.07(d,J)3.6hz,2H),4.51(t,低分辨,2H),3.86-3.72(m,4H),3.57-3.54(m,2H),3.42(m,4H),3.18(16H)1.80(t,低分辨,CH2,4H),1.38-1.30(m,44H),0.91(t,J)6.4hz,CH3,6H)。13C NMR(100 MHz,CD3OD)δ98.94,74.58,72.89,72.66,68.80,66.68,66.47,52.70,33.20,30.91,30.84,30.74,30.61,30.50,27.58,23.86,23.69,14.62。FAB-HRMS(M-Cl)+:计算825.6335。发现:825.6310。肛门。C44H90Cl2N2O9的计算:C 61.30,H 10.52,Cl 8.22,N 3.25。发现:C61.06,H10.45,Cl 8.17,N3.23。中央军委。(A)根据du Nuoy环法,使用21型Fisher表面张力仪测量表面张力。手动升高和降低6 cm Pt/Ir环,并在每种浓度的分析后,通过交替浸入0.1 M HCl和Milli-Q纯化水(18 MΩ‚然后在本生灯上进行火焰干燥。使用在Pyrex结晶皿中用Milli-Q水制备的溶液(25 mL)。每种浓度下B-14的报告值是混合后立即进行的10次单独测量的平均值。由于B-12的动态表面张力特性,每隔20分钟读取一次读数(在老化期间,环浸入溶液中);报告的值来自每个浓度下两次测量的平均值。随时间变化的表面张力表现为在每种浓度下的10次测量中不断增加,随后通过使用µ槽装置进行的动态表面张力实验进行量化,如下所述。(B)使用YSI 35型数字电导仪在23°C下进行电导实验。在25毫升玻璃培养管中制备每种gemini在Milli-Q纯化水中的溶液。在读数稳定几分钟后进行测量,从最低浓度的溶液开始,以避免在样品之间清洗电极。当一种表面活性剂的所有溶液分析完成后,用溶剂清洗电极,清洗顺序为:丙酮、甲醇和Milli-Q水。差示扫描量热法(DSC)。使用哈特科学差示扫描量热仪测量合成表面活性剂的转变温度。用溶剂清洗四个金属安瓿或细胞,清洗顺序为:Milli-Q水、丙酮、乙醇和甲醇;由于其他溶剂干扰了橡胶垫圈,因此仅使用Milli-Q水和乙醇清洗盖子。安瓿和瓶盖在110°C烘箱中干燥至少4小时,并在使用前立即冷却至室温。在水浴中交替加热含有2 mg表面活性剂/mL Milli-Q水的混合物,并通过漩涡(科学产品高级混合器)混合,直到形成不透明悬浮液。在使用汉密尔顿玻璃注射器将500µL等分试样注入三个样品池之前,摇动混合物;校准池中使用500µL Milli-Q水作为标准。以10°C/h的速率加热量热计,在向上扫描和向下扫描之前分别保持30和10分钟。将数据转换为热容(µJ/°C),并使用Microcal Origin软件绘制。为确保结果的准确性,对每种表面活性剂进行了三次测量。标准品二棕榈酰磷脂酰胆碱(DPPC)的41.1°C转变温度与报告的41.2°C值相对应,48进一步验证了我们仪器的准确性。动态光散射。使用Coulter N4 Plus颗粒表征仪测定表面活性剂聚集体的大小。反应杯用乙醇和Milli-Q水冲洗,并用压缩空气干燥。使用带有Whatman一次性过滤器(200或450 nm)的注射器将含有溶液的三毫升样品注入反应杯中,所述溶液的浓度高于估计浓度或表观浓度(分别为A-8和B系列),或通过稀释500µL挤出的1 mM囊泡溶液(A-10)。在20°C下,以90°的散射角进行三次单独测量,预标定时间为2分钟。a-8和a-10使用2分钟的运行时间,而B系列阳离子表面活性剂分析30分钟。选择与强度模式相反的统计重量,以更准确地描述特定规模的总体。电影平衡。使用位于水平大理石台面上的Kibronµ槽进行压力面积等温线测量。每次测量前,用乙醇和Milli-Q水彻底清洁槽和特氟隆屏障,并用压缩空气干燥。用乙醇清洗导电针,并在本生灯上进行火焰干燥。通过汉密尔顿玻璃注射器将每种表面活性剂的10微升1 mM储备溶液和CHCl3/MeOH(体积比为9/1)的混合物小心地涂抹在槽中的一层Milli-Q水的表面。在覆盖放置10分钟以允许溶剂蒸发后,屏障以4Å2/链/分钟的稳定速率自动压缩,并由仪器记录获得的压力/面积数据。每次运行后,对槽和附件进行大力清洁,并对每种表面活性剂重复该程序10次。通过将导电针插入Pyrex培养皿中新搅拌的溶液中,µ槽装置还用于量化B-12显示的动态表面张力行为;因此,监测了表面压力随时间的变化,并用仪器记录了测量结果。通过挤压制备大囊泡。使用乙醇和Milli-Q水彻底清洁Avestin Lipsofast挤出设备。将A-10(500µL,1 mM)的水混合物在5 mL圆底烧瓶中磁搅拌3 h,得到不透明混合物,该混合物通过100 nm聚碳酸酯膜挤出19次。通过水合作用制备巨大囊泡。17约1 mg表面活性剂沉积在1.5 cm(内径)橡胶O形环孔内,并与硼硅酸盐玻璃显微镜载玻片粘合。用0.5毫升Milli-Q水对样品进行水合,在顶部放置一个干净的盖玻片,并将多余的水排出,以提供紧密的密封。为了防止细菌污染,水中加入了微量叠氮化钠。允许A-10、A-12和A-18U样品在室温下放置,而A-14和A-16样品在45°C下使用安装在尼康隔膜TMD上的定制塑料孵化壳内的尼康孵化器孵化。分子模拟。使用Macromodel38软件在硅图形计算机上进行了模拟。在选择没有此类参数的琥珀*(AMBER*)之前,对海藻糖能量最小化中的低能参数数量进行了可用力场评估。在进行的25000次构象搜索中,将海藻糖分子上的环碳标记为手性中心,选择水作为溶剂。在搜索过程中,对工作集结构中的所有原子进行了比较,并选择了扭转键。搜索中使用的结构是前一次搜索中能量最低的构象。因此,通过琥珀*(AMBER*)的25000构象搜索,海藻糖结构的最低能量构象用乙酰氨基或三甲基铵基对6-和6′-羟基进行了修正。然后,乙酰基被十二烷基取代,三甲基铵基被二甲基十二烷基铵部分取代。琥珀色*使这些结构的能量最小化,导致链的全反式取向。随后,对能量最低的结构进行另一次25000构象搜索,除每条链上的第一个亚甲基外,所有亚甲基均未选择扭转键,然后搜索选择扭转键。对于A-12分子,C2-和C2′-羟基被转化为甲氧基,从而不允许与相邻葡萄糖环上酰胺的羰基发生氢键相互作用。最后,在能量最低的构象上进行能量最小化,甲氧基回复为羟基,得到如图6所示的构象。对于B-12二铵双子,对三种结构进行构象搜索,其链相对于糖部分的初始位置不同;图7所示的相同的最低能量构象导致了这三种情况。
致谢
我们感谢Jim Snyder博士和Dennis Liotta博士慷慨地为我们提供了数小时的硅图形工作站使用时间。我们感谢Jason Keiper博士在光学显微镜方面的帮助,以及Ben Cornett在宏模型建模方面的有益建议。这项工作得到了陆军研究办公室的支持。