




合作客户/
拜耳公司 |
同济大学 |
联合大学 |
美国保洁 |
美国强生 |
瑞士罗氏 |






相关新闻Info
-
> 泡沫形成的原理是什么?阴离子表面活性剂为何可以作为起泡剂?
> 超微量天平应用实例:氧化焙烧除硒火试金重量法测定粗硒中金、银含量
> 纳米熔盐形成机理、表面张力测定及影响因素研究(一)
> 不同类型的聚醚类非离子破乳剂对PPG-稀释原油界面膜性质的影响(上)
> 身体对称性和表面张力有什么关系
> 基于表面张力方法判断物质(或材料)的亲水性(一)
> 橘皮素与环糊精在油水界面自组装行为对脂质消化的影响
> 4种新型稀土双酞酞菁衍生物合成及LB膜的制备
> 粉体材料润湿接触角测量方法应用于表面张力仪
> 诱导期测定法研究NaCl的添加对碳酸锂固-液界面张力等成核动力学参数影响——过饱和度的计算
推荐新闻Info
-
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(三)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(二)
> 羟基氨基改性硅油的合成、表面张力仪测试及其在炭纤维原丝油剂中的应用(一)
> 表面张力修正系数与蒸汽进口参数的相关性
> 表面张力修正系数与蒸汽膨胀速率的相关性
> 蒸汽自发凝结数值模拟中液滴表面张力修正系数的确定方法(一)
> 融合小波分析的神经网络模型在CO₂-水界面张力预测中的优势与应用
> 表面张力——高精度玩具图案转印的“隐形基石”
> 表面张力均衡在除尘滤袋中的关键作用与革新
> 超低界面张力下重油-水两相垂直流动型态实验研究与图版预测(四)
热力学模型计算MgO-B2O3-SiO2-CaOAl2O3富硼渣表面张力(二)
来源:中国有色金属学报 浏览 1246 次 发布时间:2024-08-13
2 MgO-B2O3-SiO2-CaO-Al2O3体系熔渣表面张力模型的建立
Butler假设熔体表面相内组分与体相内组分在热力学上都达到平衡,推导出表面张力与热力学性质之间的关系(Butler方程)。本模型基于Butler方程计算熔渣表面张力,熔渣表面相和体相内组分通过熔渣结构离子与分子共存理论来确立。本模型主要假设如下:1)熔渣表面相与体相都遵守熔渣结构离子与分子共存理论,即组元结构都由简单离子、分子和复合分子组成,熔渣表面相和体相中简单离子和分子进行着形成复合分子的动力学质量平衡反应,且表面相和体相中形成复合分子的反应都遵守质量作用定律;2)熔渣表面相和体相中各组元的质量作用浓度和熔渣表面张力符合Butler方程:
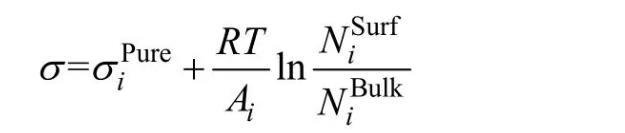
式中:σipure表示纯组元和Al2O3)的表面张力;Ai为纯组元i的摩尔表面积,其中L为校正因子,熔渣中设为1.091;N0为阿伏加德罗常数,Vi为组元i的摩尔体积);R和T分别表示摩尔气体常数和绝对温度;为组元i在表面相或体相的质量作用浓度。
根据共存理论以及上述确定的MgO-B2O3-SiO2-CaO-Al2O3富硼渣体系熔渣中存在的结构单元,定义熔渣中成分分别为。结构组元作用浓度符号表示为:
所有组元总平衡摩尔数表示为∑ni。各组元作用浓度表达式为:
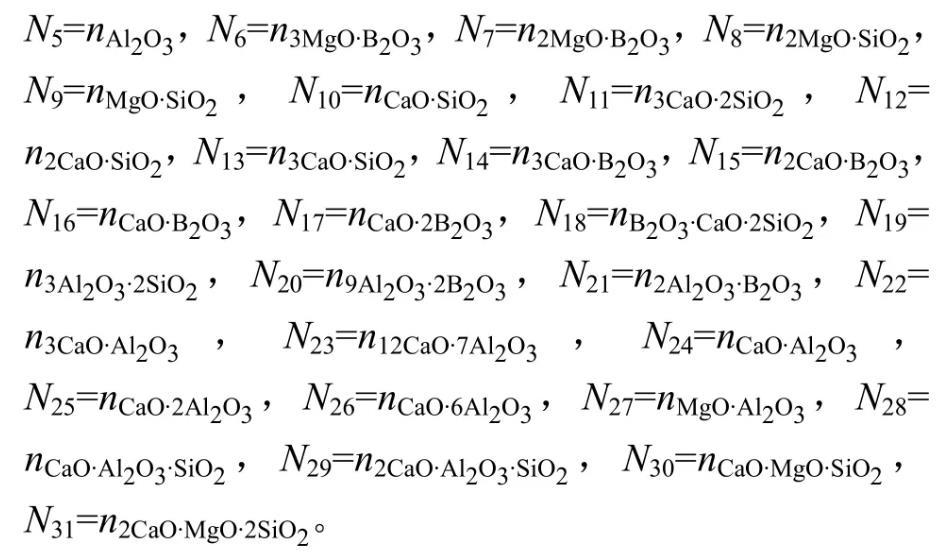
在1773~1873 K温度范围内,熔渣中各组分之间形成复杂分子的反应式及其达到平衡时的标准Gibbs自由能(以纯物质为标准态)和质量作用浓度的表达式如表1所列,其中所有反应的平衡常数可通过的关系式进行计算。
MgO-B2O3-SiO2-CaO-Al2O3渣系中质量平衡公式如下:
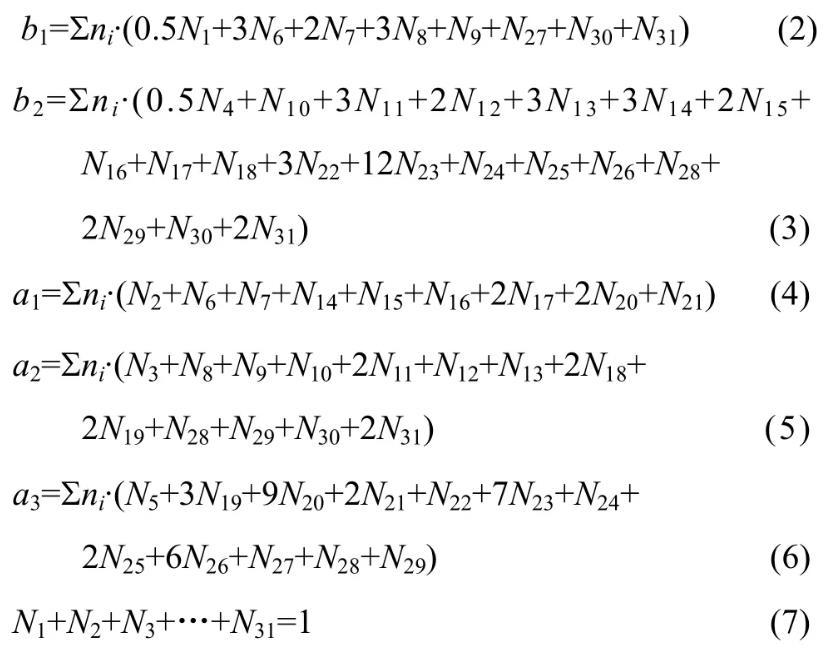
因此,由表1和式(2)~(7)建立计算MgO-B2O3-SiO2-CaO-Al2O3渣系中结构组元和离子对作用浓度Ni的控制方程,其中N6~N31由N1~N5表示出来。在一定温度下,熔渣成分代入该方程组,采用迭代法计算出所有结构组元和离子对的作用浓度。
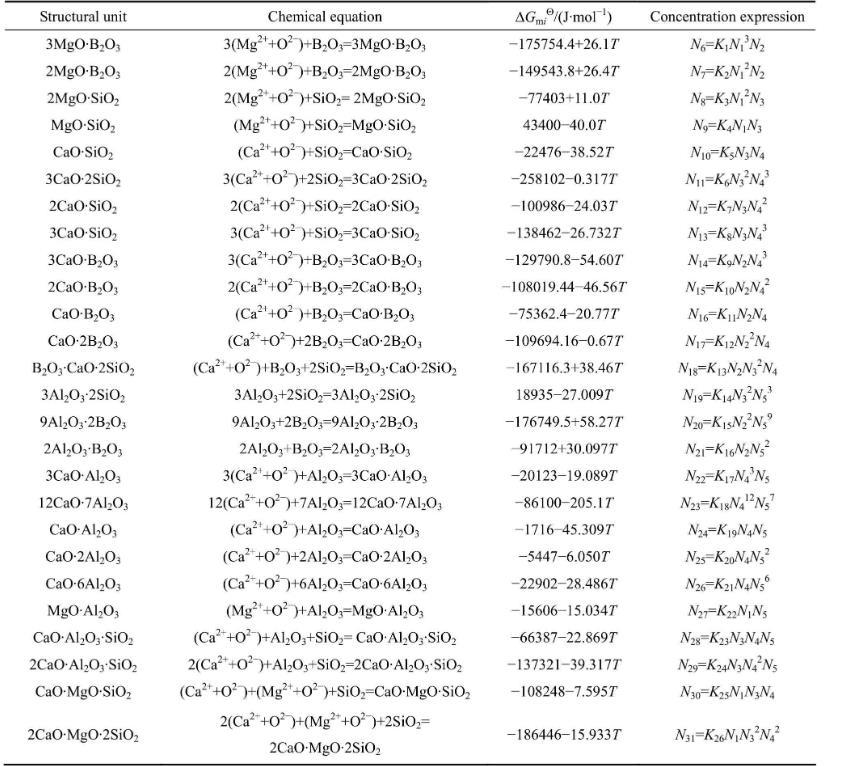
表1 MgO-B2O3-SiO2-CaO-Al2O3渣系中复杂分子的化学反应及标准Gibbs自由能和作用浓度的表达式
对于MgO-B2O3-SiO2-CaO-Al2O3体系,依据Bulter方程,其表面张力可以分别表示为
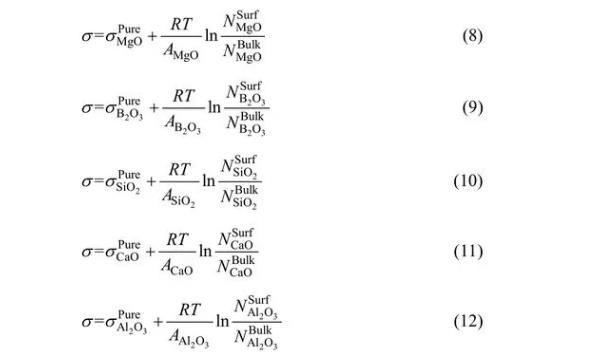
依据上述描述,建立模型可计算熔渣表面张力,其中,NiBulk可以通过熔渣成分的摩尔分数和基于熔渣结构离子与分子共存理论的形成复合分子的化学平衡计算得出。然后进一步基于共存理论和Butler方程,通过已知量NiSurf、σiPure和Ai,利用方程(8)~(12)可计算出σ和NiSurf值。MgO-B2O3-SiO2-CaO-Al2O3渣系中σiPure和Ai数据可见表2和3。
3计算结果及讨论
3.1表面张力的计算值与实测值对比
为了验证本模型计算结果的准确性,需将计算结果与文献实验数据进行对比。富硼渣相关体系中,已有B2O3-CaO体系、B2O3-SiO2-CaO体系、B2O3-CaOAl2O3体系、B2O3-SiO2-CaO-Al2O3体系和MgOB2O3-SiO2-CaO-Al2O3体系的表面张力实验数据的报道,相关体系的组分范围及温度范围如表4所列。本研究中计算了上述体系的表面张力计算值,并与文献实验数据进行了对比,对比结果如图1和表4所示。5个体系文献实验结果与计算结果总平均相当误差为9.03%。大多数熔渣的表面张力实验误差一般为±5%~10%,由此可知,本模型计算的熔渣表面张力值与实验值吻合较好。比较结果显示,B2O3-SiO2-CaO体系和B2O3-CaO-Al2O3体系偏差较大,这一方面可能与表面张力高温测量难度和精度导致的误差有关,另一方面本模型未考虑熔渣中部分组元会存在饱和现象。由于氧化物纯物质的熔点偏高,计算温度下采用的氧化物纯组分表面张力数据由已有纯物质的实验数据进行合理的外推得到,这些也可能对模型计算结果带来一定的误差。
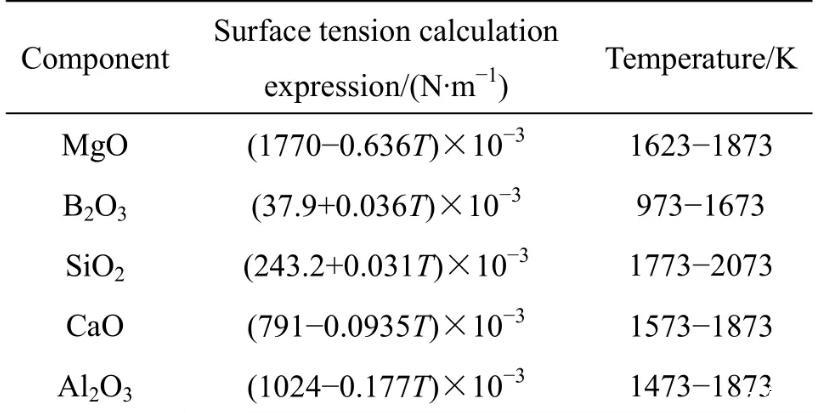
表2纯组元表面张力与温度的关系

表3纯组元摩尔体积与温度的关系
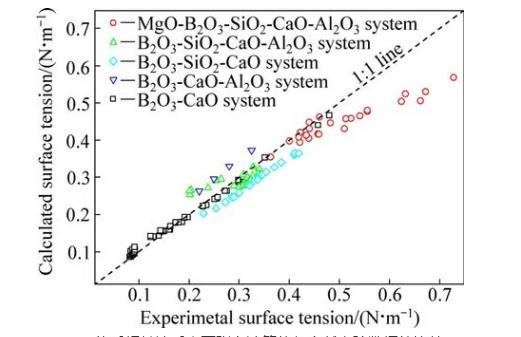
图1 MgO-B2O3-SiO2-CaO-Al2O3体系相关渣系表面张力计算值与文献实验数据的比较
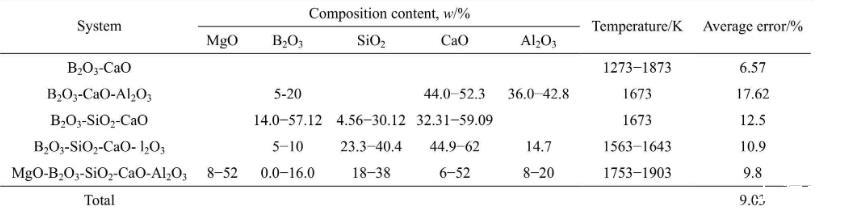
表4富硼渣相关实验渣系组分、温度及相对平均误差
热力学模型计算MgO-B2O3-SiO2-CaOAl2O3富硼渣表面张力(一)